Pro-1
Pro-1 is a fully open source reasoning model (8b and 70b) trained using GRPO towards a physics based reward function for protein stability.
It takes in a protein sequence + text description of the protein + past mutagenesis results, reasons over the information given, and proposes modifications to improve the stability of the given sequence.
Background
Protein instability is a common problem in biology where proteins unfold in harsh environments, such as higher temperatures or acidities. Instability can hinder applications in drug development, biomanufacturing, and synthetic biology, where stable proteins are crucial for therapeutic efficacy, industrial enzyme performance, and engineered protein designs.
Wet lab directed evolution works well, but is incredibly resource intensive and cannot possibly explore sequence space efficiently. Directed evolution is also prone to getting stuck in local minima. For the average 100 amino acid sequence, there are 10^50 times as many possibilities as there are atoms in the observable universe.
Fortunately, many researchers build intuition about a variety of properties when designing their protein sequences and can instinctively optimize their sequence towards some desired property in the same way that an engineer can finetune an engine to improve performance. For example, after the discovery of CRISPR, scientists were struggling to have the CRISPR complex enter the nuclei of mammalian cells. It wasn't until Feng Zheng's lab had the idea to attach a nuclear localization sequence to the end of the protein that CRISPR could be successfully delivered in human cells.
This type of intuition is built over years of understanding scientific literature and experimentation. But what if we could build this intuition in a language model by allowing it to self play using a physics based reward signal?
Why a Language Model?
In addition to the rational design argument, there are many appeals of a language model compared to the black box protein language models and bindcraft style sequence optimizations.
- Language models see a massive corpus of scientific literature during training, and therefore have a strong prior to propose useful modifications.
- Every modification is highly interpretable and can be directly questioned by a researcher (good interactivity).
- There is greater potential for creativity.
- Language models are super flexible. I can prompt the model with whatever I want: descriptions of experimental results, papers, PDB structures, secondary structures, etc.
- LLM's are on a rising tide–as general capabilities improve they should also improve for biology specific tasks.
But most importantly, it's just good fun.
For the base models, I used Llama-3.1-8B-Instruct and Llama-3.3-70B-instruct. All training unless otherwise specified was 4 bit QLORA. GRPO was only done on the 8B model.
SFT on Synthetic Reasoning Traces
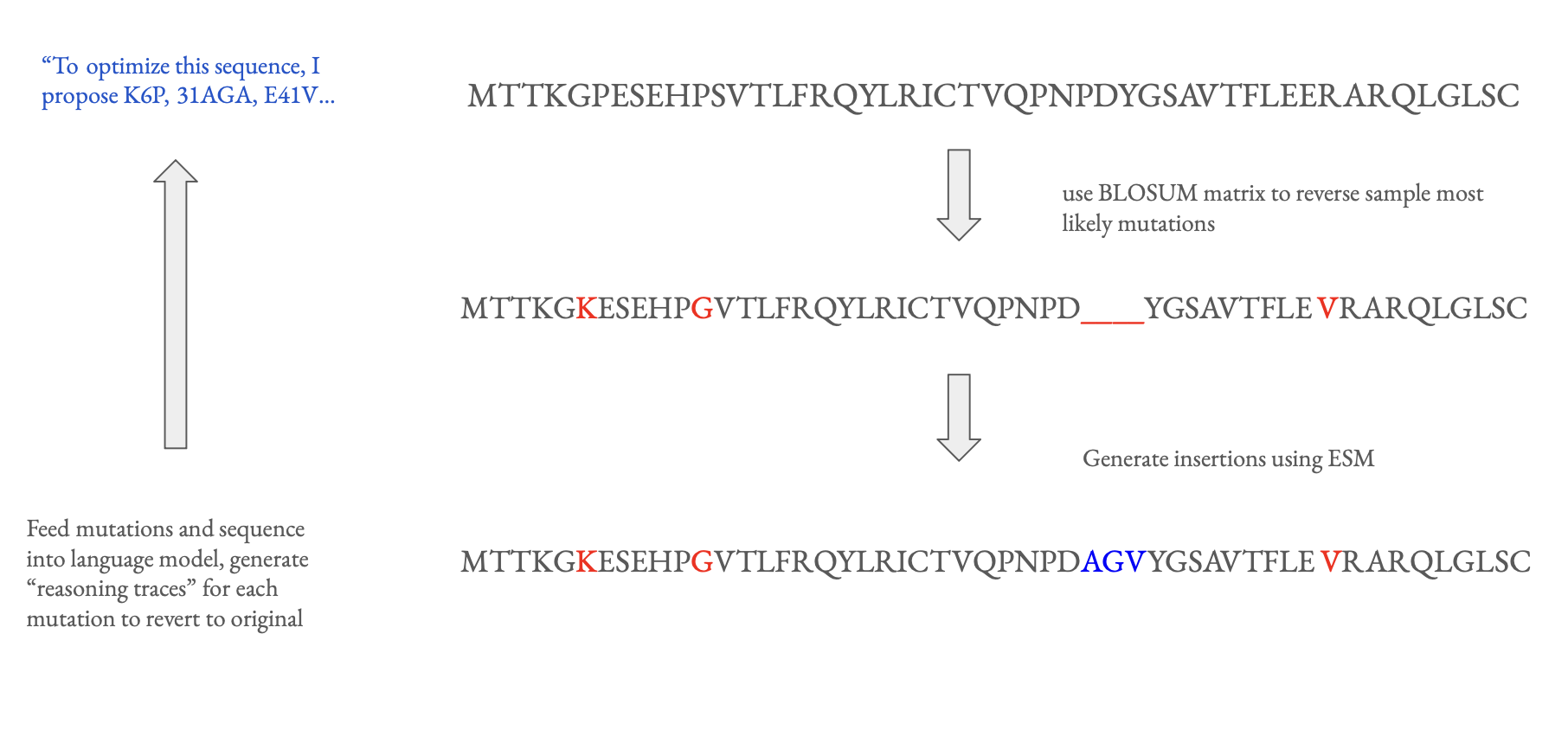
To get the model in the general right direction, I decided to do SFT on synthetically generated reasoning traces before the GRPO. With the argument that evolution is the ultimate optimizer, I sought to leverage the massive database of naturally occurring sequences and further expand on this by synthetically generating more data.
Using sequences from the BRENDA database, I perturbed each "ground truth" sequence using a BLOSUM substitution matrix (a 20x20 transition matrix for each amino acid based on evolutionary data) and ESM3 to generate statistically likely insertions, deletions, and point mutations. Then, using a language model, I generated reasoning for each of the mutations to go from the perturbed to recover the original sequence. The base model was then finetuned using the reasoning dataset generated.
The reasoning is generally pretty uninteresting, with most explanations being nonspecific to the enzyme given. Occasionally, the language model will reason specifically about the given model's active site or its function, but this is relatively rare compared to reasoning about the amino acid being changed or general reasoning about stability.
This method needs to be tested more but has substantial implications if it scales well, especially since bio data is exceedingly scarce. By treating existing sequences as more optimized forms, we can generate infinite optimization data by leveraging the enormity of the search space against itself.
GRPO on Rosetta Energy Function
Deepseek recently made GRPO famous with their R1 launch, showing impressive results with more efficient and stable training. For an incredible and intuitive explanation on how GRPO works, I would recommend this blog post by Yihua Zhang.
The rosetta energy function (REF2015) is a physics based scoring function used to evaluate and optimize protein structures, sequences, and interactions. It incorporates Van der Waals interactions, electrostatics, statistical potentials, and other physical chemistry terms to estimate the energy state of a structure. Most importantly, it has been shown to be a strong predictor of protein stability and has become industry standard for this task.
You may be wondering, if we have access to a closed form reward function, why not take a bindcraft style approach and simply do optimization over the sequence itself? This is a valid question and aside from the benefits of the language model above, the rosetta energy function has many terms that are nondifferentiable. If you were to remove these terms, it is unclear how transferable REF2015 is.
In the training loop, Pro-1 is given a plethora of information such as sequence, reaction mechanism, previous mutagenesis results, etc. It then reasons over the information given and proposes modifications to the sequence. The modified sequence is passed to ESMFold which generates a structure for the modified sequence, and passes this structure to the Rosetta energy function for stability scoring. From this score, a binary reward indicating stability improvement is used for the GRPO along with a simple formatting reward. I tried both a relative percentage reward as well as the absolute delta, but both of these were less stable than the simple binary reward.
By optimizing towards the rosetta energy function, the model learns heuristics about the physical world and the effects of specific mutations—without ever explicitly seeing the rosetta energy function.

Engineering Side Notes:
GRPO is much more memory intensive than SFT, I'm assuming because of the need to store more activations per batch plus the need for a reference model.
There were a few tricks I implemented to reduce the memory requirements when trying to GRPO the 70B model. Because this was a LORA, there was no need to load reference model weights into memory. Since the only difference between the reference model and the new model is the adapter, a proxy for KL divergence can be calculated using the norm of the adapter weights (not sure if this is completely true but it worked well enough).
Unfortunately, this was still not enough as FSDP appeared to do an all gather for each model.generate, leading to a number of issues since the model did not fit on a single GPU (please let me know if there is an easy fix here). Started to implement a pipeline parallel strategy for inference with one GPU dedicated as a reward server, but this was left for another day.
I also had to add a call to another language model to apply the mutations to the sequence correctly (although this was also imperfect and really should not be).
Creativity Tuning
While the base GRPO results were impressive, the model output seemed somewhat repetitive and bland, suggesting the same types of point mutations (polar aa -> nonpolar aa) regardless of input.
To counter this, I added a "creativity reward" term, where an LLM judge gives a numerical score for the novelty or complexity of proposed modifications. The judge was given examples of modifications that would be considered more creative and modifications that would be less creative. For example, deleting a protease binding site or adding a novel domain was considered more creative than modifying individual amino acids.
I also added a reward with a separate judge for specificity of the model's reasoning. This was to prevent the model from giving reasoning that was more general to the amino acids without considering the current protein it was optimizing.
After rerunning GRPO with this new reward function, the output style was noticeably different. Insertions and deletions of larger sequences with well motivated explanations were much more common, and performance slightly improved (43% -> 47% on benchmark).
In other words, the model was encouraged to be more creative and specific during RL, and it led to better performance.

Results
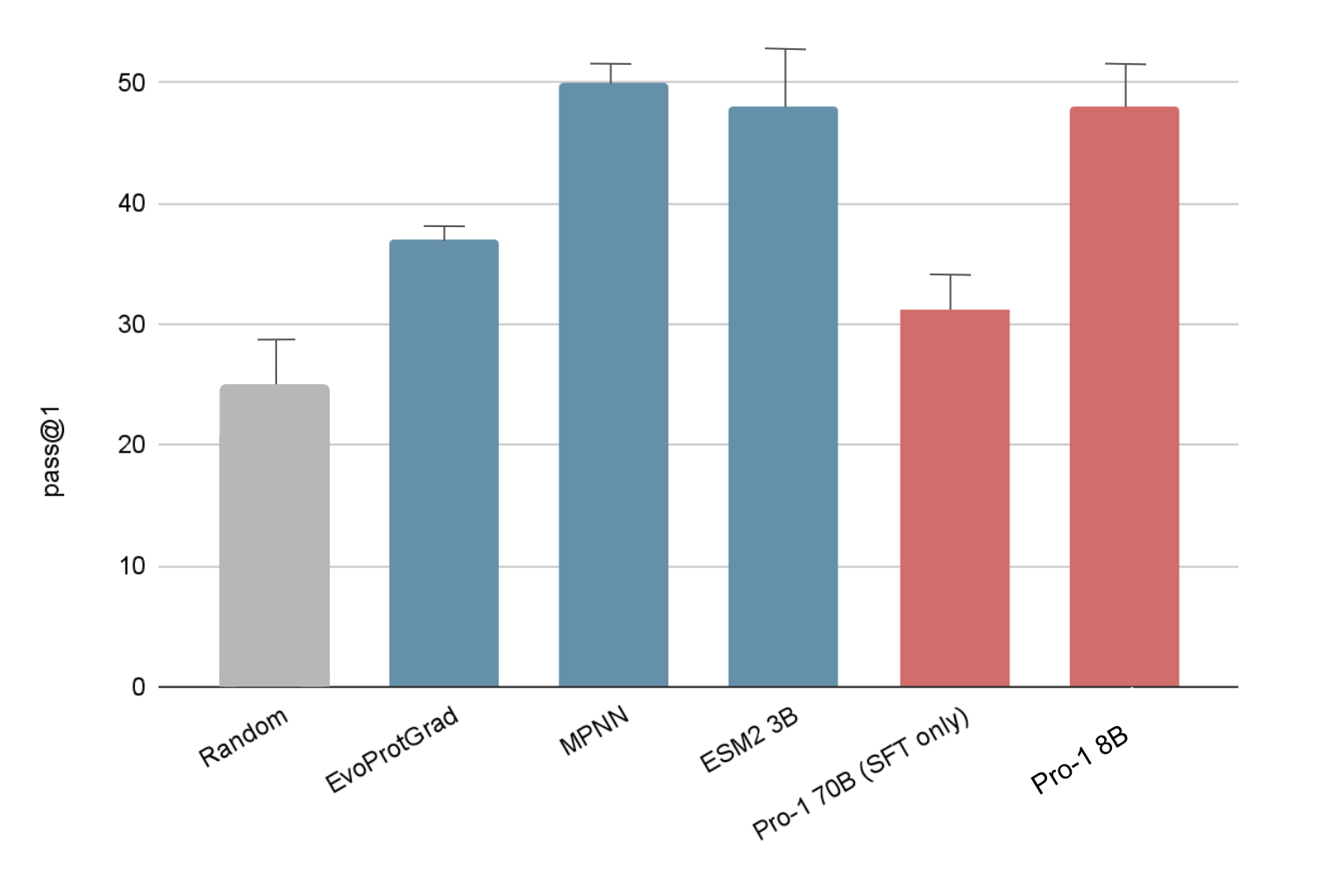
I constructed a pass@1 eval of 40 enzymes from different functional classes where success was counted if the model's proposed mutations led to a more negative rosetta energy score. The eval pipeline was the same as training, where the model would propose modifications, ESMfold would fold the structure, and the delta in energy function would determine success. With this simple GRPO, the quantized 8b param model outperformed many of the larger language models by a significant margin.
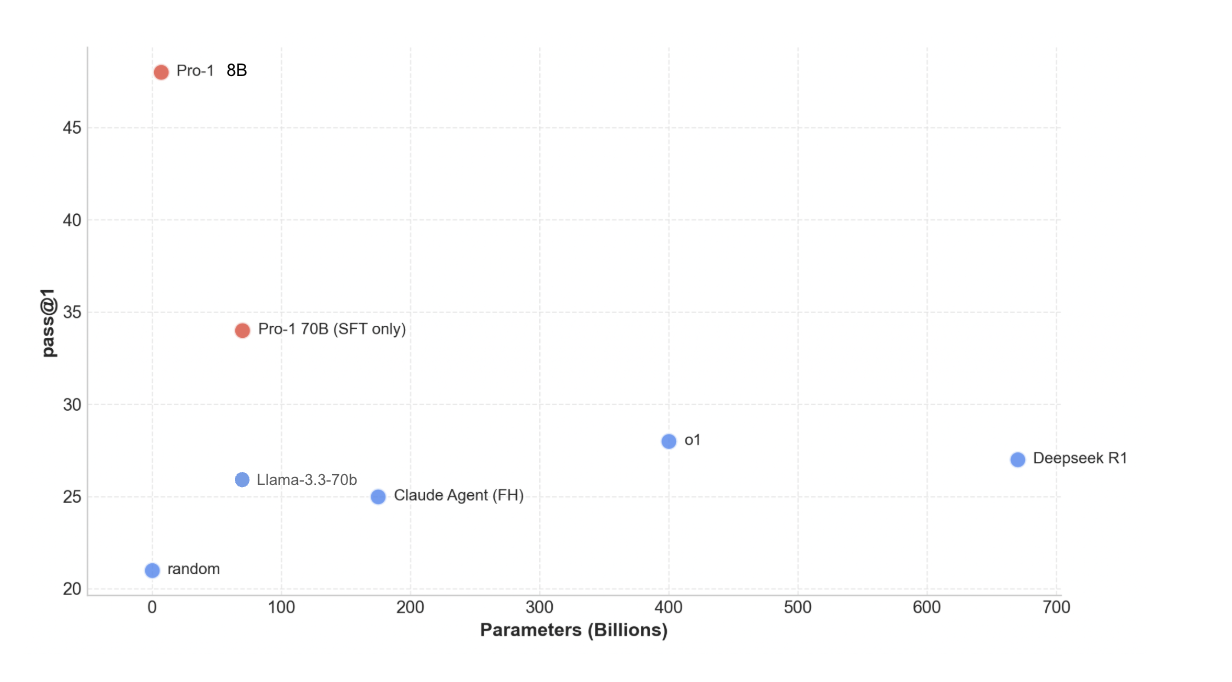
Comparing to the biology specific models required some additional engineering to do stability optimization. Since ESM does not natively do this sort of prediction, I implemented a prediction mechanism which uses the attention scores in ESM2 to select the index of the modification, then resampled ESM for the next most likely amino acid. I also implemented the Evoprotgrad method with ESM2 3B, which optimizes a sequence with respect to the internal ESM score.
Initially when testing ProteinMPNN, the model was performing exceptionally well (>70%), but was routinely changing over half of the amino acids in the sequence. To do a fair evaluation, I restricted MPNN to making 5 mutations and random, non active site locations, while fixing all of the other residues. All methods performed well, but Pro-1 remained competitive despite being trained on orders of magnitude less biological data.
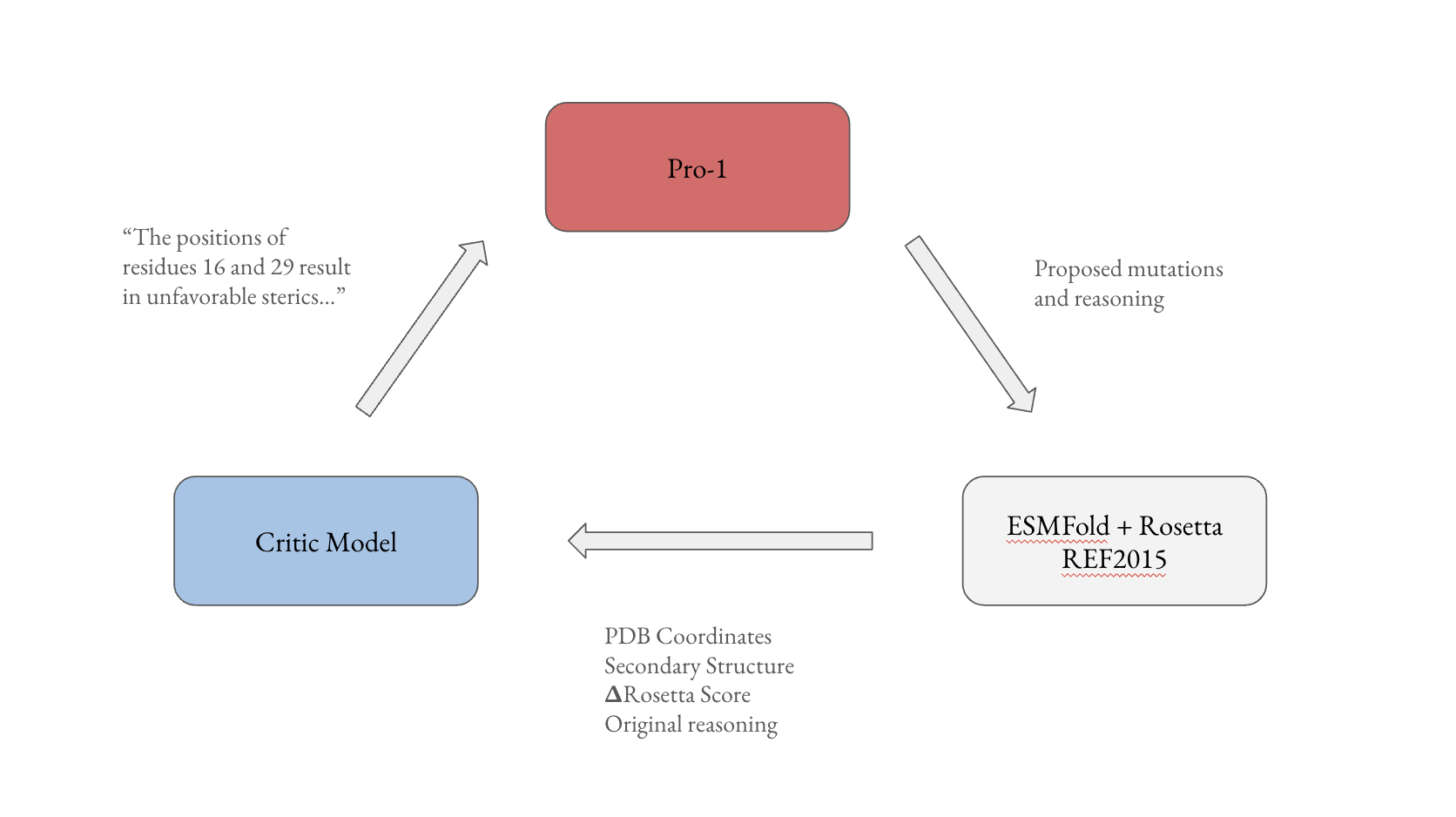
Critic Loop
In the theme of boosting test time compute, I built a loop where a separate critic model analyzes the pdb file, secondary structure, and outcome of result, and then suggests to the generator additional attempts (interestingly, much of the information I decided to pass to the model was initially requested by Pro-1). The critic examines the results and then provides specific feedback to the generator for the next iteration.
All of the above information was simply appended to the prompt and passed as text without training a separate encoder or mapper. The drawback is that passing this much context (like the entire PDB file) is clunky and requires a 1m context window. Could experiment here with passing images or structure encoding from ESM or alphafold.
After 3 iterations, the critic loop achieves a 71% pass rate, which is admittedly lower than expected given the information provided to the critic model (just resampling the model gets ~ 65%). More testing should be done here, particularly in regards to how the feedback is passed. Maybe use textgrad as inspiration?
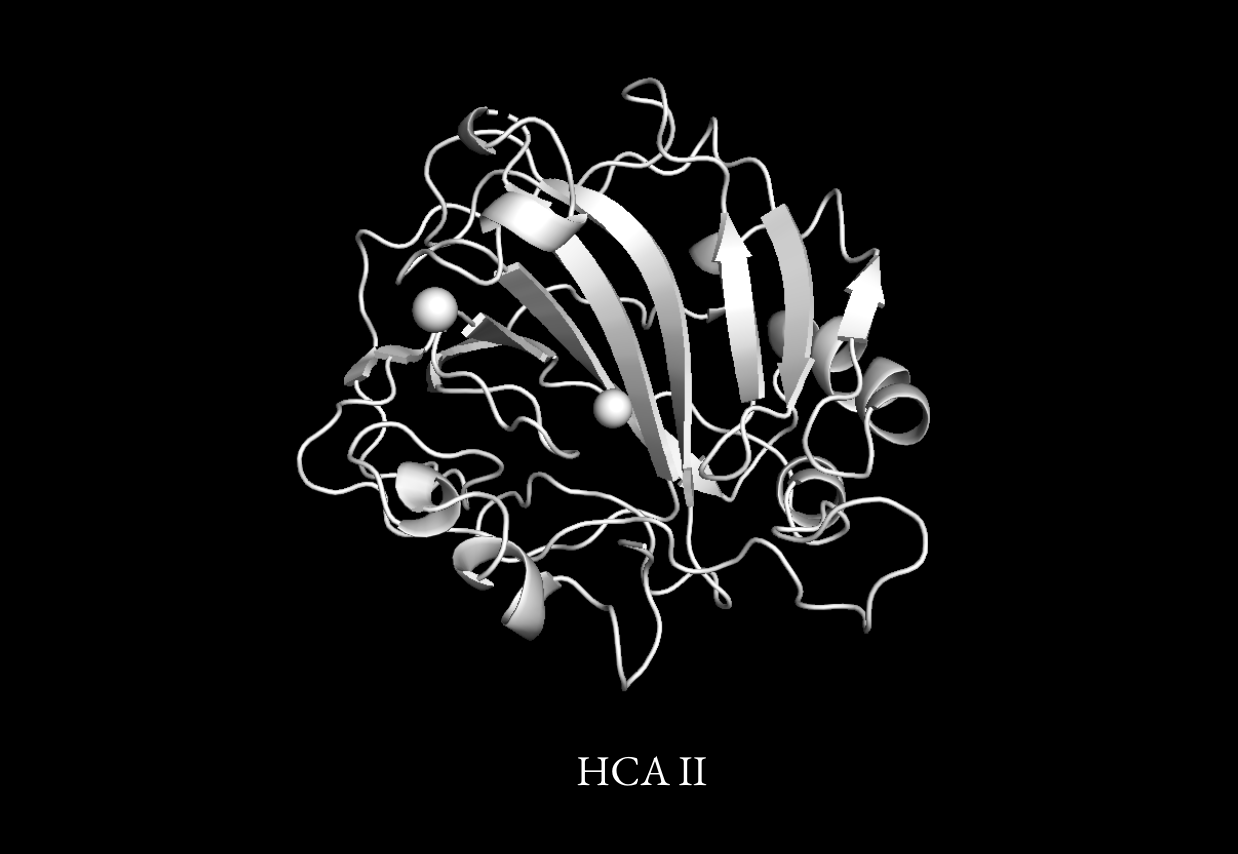
Optimizing Human Carbonic Anhydrase II (HCA II)
Enzymes have been an area of intense research for carbon capture due to their ability to catalyze CO₂ conversion with remarkable efficiency. Among these, HCA II is an exceptionally efficient candidate, speeding up the conversion of carbon dioxide to bicarbonate by a factor of 10^7.
Unfortunately, HCA II is highly unstable, limiting its practicality in large-scale industrial applications. Many industrial carbon capture processes operate at elevated temperatures, where enzymes tend to denature and lose activity. If HCA II could be engineered to maintain its function under harsh conditions, it could be integrated into industrial CO₂ scrubbing technologies and improve carbon capture efficiency by an order of magnitude.
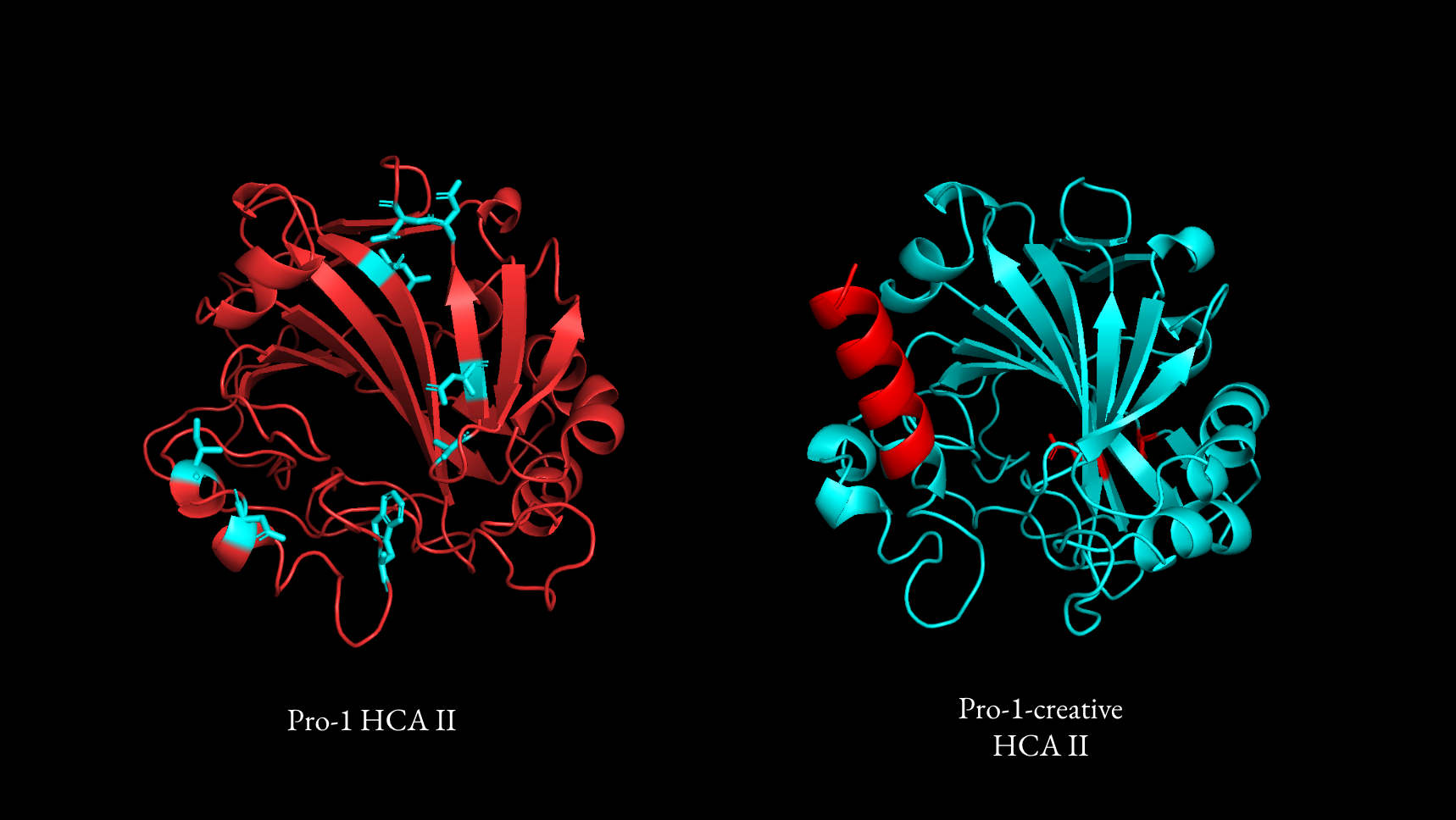
For the base model, I passed in the native HCA II sequence, effects of known mutations, excerpts from a review on the topic (Fiore, 2015), reaction mechanism, and residues that were known to be involved in the reaction. Out of 100 samples, the best proposal from the base model suggested 8 modifications, all of which were point mutations primarily motivated by reducing hydrophilicity in hydrophobic regions and vice versa.
For the creative model, I collected abstracts from two interesting papers that produced more stable forms of different carbonic anhydrases. The first, from (Sun, 2022), attached two peptides to the end of hmCA that formed a covalent bond when synthesized, leading to the cyclization of the enzyme and increased stability as a result. The other, from (Jo, 2024), added an intrinsically disordered peptide to the n terminus of taCA to act as an "entropic bristle". Its disordered nature was found to prevent protein aggregation and resulted in an overall increase in solubility, expression, and long term stability.
The creative model then reasoned through the insights from the literature provided and suggested novel modifications motivated by the themes of the papers provided. For example, in its best generation, the creative model reasoned that introducing a peptide tag would enhance protein stability and attached a helix of glutamic acids to the n-terminus. With naive directed evolution, the addition of a completely novel domain such as this would have taken an order of magnitude more cycles to discover.
Both of the top generations were predicted to be more stable with the Rosetta energy function, with the base model producing a 40% increase in stability and the creative model producing a 9% increase.
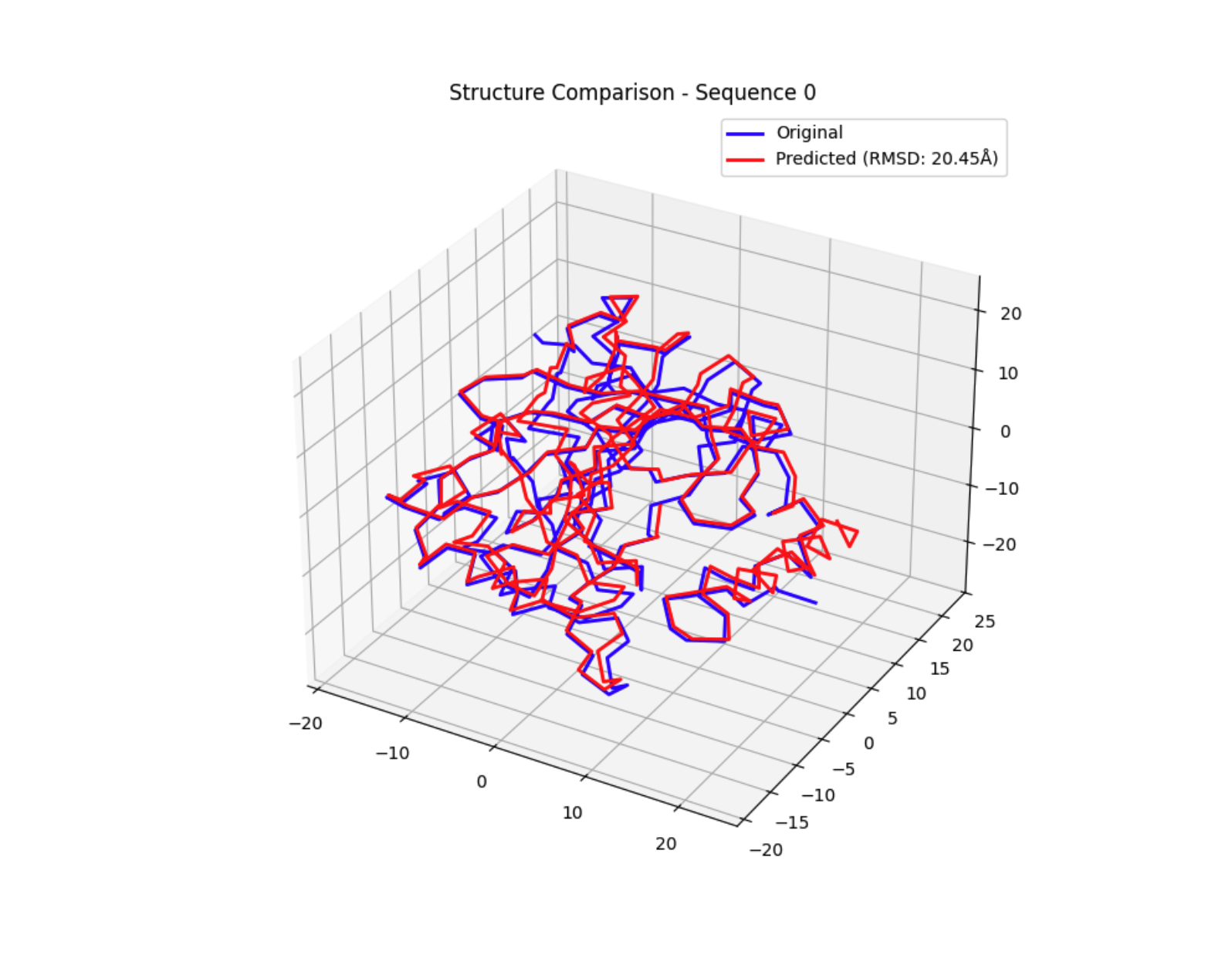
To further validate the sequences before synthesis, a quick contact mapping and normal mode analysis was performed. Also looked at RMSD between the wild type and proposed structures to preserve function. Both of the top generations showed relatively low flexibility and reasonable structure preservation.
Limitations
- The model is a preview and is often quite dumb
- can sometimes output nonsense while copying sequences.
- reasoning is often quite bland
- LLM applier can sometimes be incorrect when applying mutations to the original sequence
- The reward signal is not perfect - Rosetta energy calculations are not a perfectly accurate mapping to increased stability
- The model may just be hacking the REF2015 function, especially since the reward from the rosetta function is effectively binary.
- For example, one of the terms in the energy function is the reference energy of the amino acid in isolation, meaning that without massive structural collapse,
- The benchmark is not perfect
- A success is counted if any improvement to stability score was made.
- In experimentation, different splits have wildly different results–some runs get up to 60% accuracy. A larger, comprehensive benchmark should be made if the problem is worthy.
- A lot of stability it seems can be explained by just mutating polar residues to non polar residues, so the model may not be
- It has been difficult getting a small quantized model to output thousands of sensible tokens, so responses are often short, below 4000 tokens.
- The prompt may not have been optimized for the baseline reasoning models in the eval, and adjusting the prompt for this may boost performance
- Structure prediction models poorly predict the effects of point mutations which may have a severe effect on the reward loop.
- Metalloproteins are not well supported
- De novo design is not supported (yet)
- All references generated by the model are probably not real
- Was trained on enzyme data specifically, but should transfer to general proteins
Things That Did Not Work
- Passing secondary structure in the prompt
- Absolute and percentage reward instead of binary
- GRPO on 70b model (not enough GPUs/time)
- MCTS with constrained model outputs
- Activity reward made up of makeshift fpocket implementation and autodock vina binding scores, had very little correlation with catalytic activity
Conclusion and Next Steps
Looking forward, the biggest priority is to synthesize the model generated sequences and see if they work. Wet lab validation is absolutely necessary for a project like this, and synthesizing these sequences is the ultimate test for any model designed sequences.
Better integration with literature would also likely lead to better results. For the hCA II optimization I had to manually look up papers and extract key findings. Automating this process would better leverage the advantages of language models and likely lead to better results.
There are many other reward signals that may be useful for biology and may lead to desired outcomes. Binding affinity and activity are the two that immediately come to mind. Classifier guided rewards on immunogenicity or toxicity may also be valuable here. Curious what other interesting RL environments can be constructed across disciplines as well.
Pro-1 represents a new possibility in leveraging language models for scientific discovery. With strong reward signals, language models can reason over complex scientific tasks and serve as sources of innovation.
If you would like to contribute or have any feedback, don't hesitate to reach out. This has been my pet project over the past 2 months and there is always room for improvement.